At the intersection of neuroscience and artificial intelligence there is a popular trope that marvels at how our brain can accomplish intelligent functions with just 20 watts of power supply. Whereas current implementations of AI require the output of an electric power plant. If only we could emulate the functions of the brain in technology, we might be able to save many log units of power needed for artificially intelligent systems. Here I revisit this argument.
When people wax poetic about the marvelous faculties of the human brain, they typically imagine Einstein, Mozart, and Tiger Woods all wrapped into one person. However, these are different brains, and the combination of these skills has never appeared in the same brain together. Moreover, each of these individual brains is a one-in-a-billion outlier, and it will be difficult to learn anything useful from such extreme examples that can’t be replicated.
Meanwhile, most real human brains, most of the time, just vegetate. People can barely make it through the day without stumbling and tripping. That’s because while they cross the street, they have to look into a tiny window that they pulled from their pocket. The tiny window has flitting images on it. Those images make the human sad or happy. They determine what the human thinks and talks about the rest of the day. They can change the humans’ actions, in rare cases even lead them to kill each other.
Let’s review some of the claims for human intelligence and how they might relate to machine intelligence:
We can do math. That’s just not true. The vast majority of humans, when prompted on this topic will say “Well, I’ve never been good at math”. Even the ethereal brains of Math Olympiad winners are now getting serious competition from AI systems.
We have language. Well, that claim to uniqueness is over. Large language models can now communicate with people or each other using human language entirely. Optionally, they can translate from one human language to another along the way. Of course, if they use their own digital language, they can communicate much more effectively.
We can drive a car and load a dishwasher. The robo-taxis cruising all over San Francisco tell us that machine driving is a solved problem. Curiously, the robots imitate human behaviors that were not designed into them, such as honking at each other while fighting for a space in the parking lot. Regarding loading the dishwasher, this problem is still unsolved, mostly because it hasn’t been a priority. As a rule, the people who make decisions on technology development are not the people who load and unload dishwashers.
Which gets us back to the claim that the human brain can do all these things much more cheaply in terms of energy consumption. Is this really true? Imagine trying to replicate the functions of ChatGPT using human brains. I’m talking here about a ChatGPT model that has been fully trained and is now servicing requests for information in what has come to be called “inference mode”. Similarly, I’m allowing human brains that have been fully trained and educated and are similarly answering queries from other humans.
ChatGPT was trained on many millions of books in addition to some twaddle from the internet. As a result it acts like an expert in a million different fields of human endeavor: the sciences, technology, arts, history, culture. Today it operates about at the level of a PhD student, and the quality of its output has been steadily improving. Just like human PhD students, it occasionally makes amusing mistakes.
What would be needed to implement this core function of AI with human brains? Obviously, you would need many human brains because each of us is a PhD level expert in at best one tiny subfield of knowledge. So let’s suppose that you need 1 million human experts to cover the full range of topics. In addition, you probably want a management level, to triage the requests for information and find the correct human expert to answer them. Also, you need some services, like fast-food joints and cleaners, to keep these humans alive. Conservatively speaking, replicating ChatGPT with humans would require a medium-sized city of about a million people.
How much energy does that city consume? You might be tempted to just count the 20 watts of energy that each brain uses, but that would be a mistake. The brain is inside the body and relies on the body for everything, including the communication between brains via language. So we have to support the whole person, not just the brain. If we build that city in the United States, a human consumes about 10 kilowatts of primary energy. So running ChatGPT made from human brains would require about 10 gigawatts. By comparison, the power consumption of ChatGPT inference is estimated at ~10 megawatts, a factor of 1000 smaller. And we haven’t considered the speed of the service: ChatGPT handles about 1 billion queries a day. This would be a tough challenge for the system made of 1 million human brains.
To conclude, the human brain really is not a useful yardstick for artificial intelligence. With a few remarkable exceptions, human intelligence is nothing to write home about. And present-day AI systems already exceed human performance in many domains by many log units and for less power consumption. Looking to the future, of course it is worth contemplating new implementations of artificial intelligence. Remember that the first flying machines we invented were balloons, and that’s not the method we use for flying today. The first large language models are only a few years old, and it stands to reason that entirely different engineering solutions will emerge down the line. But regardless of the choice of technology, the future is one where machines run the world with an affordable power supply.
A small fraction of that power will go to keeping the humans entertained with flitting images on the tiny window.
by Markus Meister, Kyu Hyun Lee, and Ruben Portugues
Biology has rapidly become a quantitative science, with mathematical methods infusing all aspects of the scientific process: from the design of instruments for observation, to processing the resulting data, to inference and modeling for the extraction of knowledge. Unfortunately higher education in Biology has not quite kept up with these developments.
Most undergraduate programs include no mathematical training beyond calculus. Often times, Biology is still advertised as a route to a science career that avoids exposure to higher math. As a result, a typical Biology graduate is not prepared to follow many high-profile articles in Biology journals that use advanced mathematical methods. Even appreciating the basic phenomena of Biology often requires some mathematical sophistication. Take, for example, the Nobel-winning discoveries surrounding the mechanism of mutation (Luria-Delbrück), communication between neurons (Hodgkin-Huxley), or circadian rhythms (Hall-Rosbash-Young).
True, certain sub-disciplines in life science have had a tradition of mathematical treatments, for example population biology, or theoretical neuroscience. Typically, this mathematical foundation is limited to a subset of methods, and PhD students choosing those subjects will have to learn the associated special tools. Would it not be preferable for students to be equipped with sufficient mathematical background by the time of their first degree, and before they choose a particular discipline?
To accomplish this, we have been teaching a mathematical methods course to advanced undergraduates and first-year graduate students in the biology departments at Harvard, Caltech, and TU München. After a few years of experience with this, the faculties at Harvard and Caltech made the course a requirement in life science graduate programs. Based on those teaching efforts – and over about twelve years of gestation – we wrote this book, now out at MIT Press. See here for a table of contents, sample chapter, and other materials.
What makes this book “for biologists”? After all, mathematics is the same regardless of application area. We see our special contribution here in three ways:
• The choice of areas. We focus on three subjects that are fundamental to life science: linear systems; probability and statistics; and nonlinear dynamics. Vice versa, we intentionally chose to ignore others, such as group theory, which you would find in a corresponding book for physicists.
• The style of presentation. We treat the mathematics rigorously, but with an emphasis on practical uses, rather than theoretical abstraction and completeness.
• A broad sample of applications drawn from different areas of life science, illustrating how these methods work in practice.
This book can accompany a one-year mathematical methods course, but we have also given shorter courses covering a single semester or 10 weeks, based on appropriate selections of the material. Naturally the book also serves as a guide for self-study. And as a reference work to keep on the shelf after you have completed the course.
We also want to build a community around the book with you, the readers, using the associated web site, for example:
• The web site offers value-added materials, for example sample curricula, and the code for generating every figure in the book.
• The book contains many exercises, but no solutions. We invite student readers to produce such solutions and we will publish the best ones on this site with author credit.
• Readers can suggest further topics or new chapters, especially for the sections on advanced topics and applications. As we develop new material, it will be openly available here.
• We rely on readers to spot the inevitable errata and suggest corrections.
Most of all, we hope that this book project will help Biology students to tackle problems they might otherwise have avoided, and to bring their studies and research to a new level of quantitative richness.
A talk presented at the Symposium “Vision and Mind” University of Cambridge, Dec 2022
In late 2022 I was invited to a symposium in honor of Horace Barlow, who had passed away that year. It was a wonderful occasion, the arctic cold of Cambridge UK contrasting with the warmth of Miranda Barlow’s house and the fellowship of the guests that had come from around the world. Here is an approximate transcript of my remarks, remembering Horace through his scientific papers.
The scientists to whom I owe the most all passed away recently. Two of these – Howard Berg and Denis Baylor – I talked with every day for several years of my life. Horace Barlow, on the other hand, I met only twice at conferences. I really know him almost exclusively through his writings. So I won’t have any personal anecdotes to share, but instead want to focus on the scientific papers that left such a strong impression on me. Obviously this is but a small selection.
Looking back at my notes, I first came across Horace’s legacy as a beginning graduate student. This was perhaps the first term paper I wrote in graduate school, trying to mix some ideas from a course about Shannon’s information theory and another on neuroscience. The question was how much information compression the retina can achieve for the visual image – something that sounds familiar to many in this room. After this course I went on to measure cosmic rays in interplanetary space, and then wrote a PhD thesis on how bacteria swim. But ten years later I found myself as an assistant professor, building my research around the topic of this paper, namely information processing in the retina.
Let’s take a look at Horace’s first foray into this area. His 1953 paper on lateral inhibition in the vertebrate retina appeared in the same month as Steve Kuffler’s. These two giants of neuroscience reported what we now understand as the same phenomenon. But they came to the subject with very different styles. Steve reports the study in a workman-like and systematic manner: “first we did this, then we did that”. Horace, instead, starts the paper with a long disquisition on what one ought to expect to find, before even doing the measurements. He takes issue with an earlier report by Hartline that ganglion cells have large receptive fields with a lot of overlap. This would mean that nearby ganglion cells almost always report the same message about the image. That offends Horace’s sense that the visual system should be efficient, and so one needs to look into this with better methods. All this happens before he reports any results. I wonder sometimes how many readers fully appreciated these introductory paragraphs at the time. Obviously Horace is concerned with high-level principles behind sensory systems, but what exactly are those principles? We will get to that in a little while.
Among other things, Horace demonstrated that light has different effects depending on where it falls on the retina. A small spot of light close to the recording electrode produces a strong burst of spikes. But if you add another side spot at the same time, the response is much smaller. This was termed “lateral inhibition”. Clearly the ganglion cell doesn’t just sum light from a large receptive field, as Hartline had proposed. Instead it subtracts light in one region from light in another region. This confirmed to Horace that the retina performs much more interesting processing than previously thought. In particular, a uniform stimulus, which is a very common pattern in nature, produces only a weak response, and the retina mostly signals deviations from that expectation.
As is customary in Horace’s papers, there is also a glimpse of the future. In the last paragraph of the 1953 paper, Horace extrapolates from the reported measurements to what the retina of the frog might do with an important ecological stimulus, like a moving fly. He thinks a moving object should produce a wave of activity among ganglion cells that rides ahead of the object’s true position. This seems counterintuitive, given the response delays in photoreceptors, but is in fact exactly what happens. Some decades later, we were able to show that by recording simultaneously from an entire population of retinal neurons.
Here we come to a hugely influential article from 1961. It is published as a book chapter, so you can’t actually find it in the common literature databases. The paper also has no figures. It is all about concepts. But it uses figurative language, as illustrated by these snippets.
Horace asks “why” the brain works the way it does, not “how” it works. To me this was a refreshing revelation. When I wrestled with this paper, I had just started my professional life at Harvard in the department of Molecular and Cellular Biology. Among my colleagues it was considered taboo to ask why a biological object was built the way it was. You were to focus strictly on mechanism, and ask “how”. The “why” questions just led to fuzzy thinking that could never be tested rigorously. Better left to the people in that other Harvard department for Evolutionary Biology. I have to think that Horace experienced similar pressures early on. The 1960s saw the rise of molecular explanations in Biology. And the Cambridge department was under the heavy influence of Hodgkin and Huxley and their success at bottom-up mechanistic modeling. So I suspect that he is reacting to those trends in this paper.
So what did Horace suggest is the purpose of sensory processing? The article considers a wide range of explanations, but eventually focuses on these ideas: 1. The first purpose is to search for patterns in the sensory data that are of likely interest. For example the image of a face or a familiar object. Such patterns by necessity reflect redundancy in the data. This means that some of the pattern can be predicted from knowing another part. For example, faces are symmetrical, so you can predict the right half from knowing the left half. Redundancy also has a strict technical meaning in the context of information theory. 2. The second goal of the sensory system is to reformat the signals so these special patterns are encoded very efficiently with just a small number of spikes. In the course of doing that, the redundancy gets reduced. There are fewer patterns left in the output signal than in the input. So the core hypothesis of this paper is often quoted as “redundancy reduction”.
This paper has an interesting citation history. For about 30 years, there was no resonance in the community. Then suddenly around 1990, the citations start taking off, and popularity has grown every year since. So if you are a young student wondering what will be the next hot topic in neuroscience, I suggest reading Horace’s papers from 30 years ago for suggestions. He really was 30 years ahead of his time.
But where could an enterprising young student find that paper? Some of them are in remote book chapters and hard to find. Trinity used to have a web site that archived all of Horace’s writings, but somehow it has gone defunct. I think it’s time to rebuild and advertise such an online resource.
So what actually happened in 1990? That’s when the power of Horace’s ideas was recognized, and it produced an explosion of work on efficient coding theory. Today this framework forms one of the few normative principles in theoretical neuroscience. The papers here are picked from vision research, but the same ideas can explain a remarkable range of structure and function in other sensory systems. It appears that the efficient use of neural impulses really is a important design criterion for the nervous system.
Speaking of design criterion, let me return to an earlier paper from 1952. Here Horace wonders about the optical design of the insect eye. Insects, as you know, have an eye with many hundreds of little facets.
Each facet is a tiny lens, and it looks out into just one direction of space, and reports the intensity there. So how big should such a facet be? If you make the lenses smaller, you can obviously fit more of them onto the surface of the eye, so the directions in space get sampled more finely. You might think that is good for spatial resolution. But if you make the lens too small then you get bit by the diffraction of light: A small lens collects light not just from one direction, but from a whole cone of light, and the acceptance angle increases as wavelength divided by lens size. So there’s a tradeoff between sampling density and this directional smear. The optimal setup is when those two angles are about the same, and that suggests the lens size should grow as the square root of the eye size.
With this prediction in hand, Horace went to the local Museum of Comparative Zoology, opened some drawers with species of hymenoptera, and measured their eye size and facet size. And in fact, the eye size (R) and facet size (D) vary together in just about the predicted relationship. An interesting case of optimal design in Biology.
This is another example how asking the “why” question helps in understanding. We no longer have to remember the facet size of all thirty of these insect species. It’s sufficient to know the principle along which the eye is designed. If you’re trapped on a desert island, you can recreate this argument by drawing lines in the sand, and easily reproduce all these facet sizes.
Incidentally, this beautiful problem seems to get rediscovered periodically. Feynman (1963) presents a derivation in his Lectures. In typical physicist fashion, he looks up the dimensions of just one insect eye, and is happy to declare victory. And there is also a “prediscovery” in a paper from A. Mallock in the 19th century.
Finally, this drawing from Kuno Kirschfeld reminds us that even the most beautiful theory in Biology has a limited domain of validity. If we extended Horace’s formula for optimal eye size to the spatial resolution of human vision, this is how we would end up. Obviously Nature has hit upon an alternative design for the eye of vertebrates, using a single large lens shared by all the photoreceptors, which fits into a much smaller package.
During the early 1960s, Horace spent time in Berkeley, where he worked with Richard Hill, Gerald Westheimer, Bill Levick and other vision scientists. Westheimer tells an anecdote that Horace was on an exploratory trip to Berkeley, when they visited Hill in his laboratory, who was recording from some cells in the rabbit eye. Horace was intrigued and waved a flashlight at the ceiling. When he waved it one way the neuron fired, but when it waved back the cell stayed silent. Apparently this was the discovery of direction selectivity in the retina. Here they plotted the receptive field of a retinal ganglion cell. When a small spot of light moves from bottom to top the cell fires a lot; when the spot moves the opposite way there is silence.
Parenthetically I’m not sure we could make such a serendipitous discovery anymore. In those days, you recorded from one cell at a time, and the spikes from the electrode were fed to a loudspeaker in the room. So you could hear exactly what the neuron signaled while you explored all kinds of stimuli. These days people record from 1000 cells at the same time, and you can’t put them on a loudspeaker. So you find out what they did only the following day after a lot of analysis, and you can’t change the stimulus until the next recording.
Barlow and Levick proposed 2 models for how this might work – note that’s twice as many models as in a typical neuroscience paper. And their analysis strongly favored one of the two, in which there is an asymmetric inhibitory connection. After much back and forth over the past half century that idea still stands.
We now know that the sampling elements B and C are bipolar cells, the summing element is the ganglion cell, and the delayed inhibitory signal comes via an amacrine cell, called “starburst amacrine”.
Of course the important part of the model is the asymmetry: the amacrine cell should send its signal only from right to left, but not from left to right. This was finally verified 50 years after the proposal by painstakingly reconstructing the synaptic connections in the retina through electron microscopy. In fact, a ganglion cell that prefers upward motion gets its inhibition only from starburst cells in the downward direction.
This 1972 paper comes closest to a manifesto, in which Horace lays out his deep beliefs about how the brain is organized. It has had an enormous influence on by now several generations of neuroscientists. Each of us can read this and find something different that catches your attention. In fact every time I read it I find new insights.
Horace wanted to draw some conclusions from what – even 50 years ago – was a profuse wealth of data about the brain. What have we learned from all this at a high level? And he sets out to summarize this by a set of principles that are consistent with the available evidence but go substantially beyond it. He presents these ideas in terms of dogmas. I suppose he was inspired here by the central dogma of molecular biology. But the statement was also intended to challenge people and invite critique. I won’t delve into the dogmas in detail, but I think these are three ideas that have had a great deal of resonance: First Horace says you need to understand the system at the level of single neurons; not at the level of molecules; and also not at the level of brain regions. The essential action is close to the single neuron level. Second, the sensory regions of the brain try to represent what is out there using the minimal number of active neurons. As one moves from the periphery to more central regions, the fraction of active neurons gets smaller and smaller. Third, as the final result of this, he suggests there may be neurons whose firing signals a rather complex event, something on the scale of a word, that forms a substantial component of our perception. A picture is worth 1000 words, so he suggests there may at any time be 1000 neurons that represent the content of our visual perception.
Some 30 years later, this idea got a great boost, through the discovery of single neurons in the human brain that are selective for high-level concepts, like Halle Berry. This neuron in an epilepsy patient fired in response to pictures of Halle Berry, her name in writing, or a picture of cat woman. But not when presented with all kinds of other actors or objects. Not surprisingly, Horace’s manifesto is the first citation in this report. This has turned into a very lively area of human neuroscience.
Finally, Horace insists that such principles should be stated in such a way to be falsifiable. And that, in fact, disproving his claims would be a sign of progress. Another attitude that has fallen out of fashion with current authors.
So let me close then by summarizing why I find Horace’s writing so inspirational.
For one, his papers “subtract from the literature”, in the sense coined by Selig Hecht. They can reduce a jumble of facts reported in a flurry of publications to a simple principle that ties those facts together. Interestingly, Selig Hecht failed in this particular instance, whereas Horace more often than not succeeded.
To accomplish such a reduction, one obviously has to look beyond the certified data and speculate a bit. Horace was always on the lookout for such overarching hypotheses, even – as we saw – in preparation for experiments, not as an afterthought. And he had no fear of missteps along the way.
Finally, they are such a pleasure to read, because of the straight-forward writing style, free of any obfuscation. Every time I read one of Horace’s papers I find a new nugget in it, because it connects to something I happen to be involved with at the moment.
With that let me thank you all for your attention, and I so look forward to the celebration of Horace’s work at this meeting.
About a year ago, the life science journal eLife changed its approach to scientific publishing (see details here). The goal was to focus the activities of the journal on peer review. Perhaps the most consequential change was to abandon the accept/reject decision. Instead, eLife reviews get attached to the author’s preprint, along with a brief editorial assessment that rates the manuscript along the dimensions of significance and strength of evidence.
In the eyes of some scientists at least, this change diminished the reputation of the journal. There was a sense that by leaving the decision to publish up to the authors, the journal would open itself up to all sorts of low-quality content. A few days ago, eLife published a self-study about the first year of experience with this model, and it is well worth a look. Here I want to add a few personal comments, based on my experience as an author and as a reviewing editor:
There’s a widespread belief that eLife’s new model is to “publish everything”. That’s just not the case. In fact eLife’s new model is just as selective as the old model: about 1/3 of submissions get sent for review.
The decision whether to review a manuscript is typically made by a group of 4 researchers: a senior editor and 3 members of the board of reviewing editors. In my practice, I read the manuscript with enough care so I could start writing a review. If I take the manuscript on as reviewing editor, I will be one of the reviewers.
One of the questions I ask myself during triage is: Would I recommend this manuscript to my students for a journal club? Because all eLife submissions are available as public preprints, this is not a theoretical question. A journal club can then be a good start to a review.
By the time a manuscript gets reviewed, it has typically been seen by 6 researchers: the above 4 and 2 additional reviewers. The reviews and a short editorial assessment get attached to the published preprint. The authors can choose to (a) do nothing further with eLife; (b) publish the current version under eLife’s banner; (c) revise the manuscript and ask for a second round of reviews.
Here are some things I like about this process, compared to many conventional journals:
The decision to review is made entirely by fellow academics, 3 of whom are chosen from a panel of hundreds for their expertise in the author’s field.
The reviews are published and, optionally, signed. This leads to a healthy degree of civility, enforced where needed by the reviewing editor.
As a rule, eLife reviews don’t tell the authors what additional experiments they should do (and what the outcome should be) in order to get published. The authors design their research report, and reviewers assess it for what it is.
The reviews are published alongside the preprint regardless of the assessment of the manuscript. Most conventional journals don’t publish reviews at all, or only if the manuscript is “accepted”. In all other cases the reviewers’ work ends up in the trash, never to be seen again.
The assessment of a manuscript is nuanced, not binary (accept/reject). It is expressed in the editorial assessment, with details in the reviews. Users can scan eLife’s web site for title/abstract/assessment and decide what is worth reading.
And a thing I would like to change:
The intent of the “new model” is to focus the journal’s product on peer review. I think we can take this one step further: There is no need to typeset the “version of record” using eLife’s fonts and page layout. In my experience that publishing step adds many weeks of delay and several rounds of proofing to correct errors introduced by the typesetters. What is the point? The paper is already published in perpetuity on the preprint server; that version is the one we reviewed; the reviews are attached in perpetuity to that version. Why duplicate the effort of typesetting and formatting? It seems like a pointless waste of time and money. Instead, we should simply list the title/abstract/assessment on the eLife web page along with a link to the preprint. That can further reduce the fee we charge to authors.
Finally a plea to you authors:
When you post your preprint, please make it a live document! It is shocking how many manuscripts on biorXiv are still built like the dead stacks of paper we mailed to the publisher in the 1980s: Pure text in the front, followed by pages of figures, followed by pages of captions, then references, and no hyperlinks between them. Really people, get with the times! It is trivial now to produce a live PDF document where figures are placed where they belong in the text; captions are below the figure; hyperlinks allow you to instantly pop to a figure, or reference, or equation; a table of contents organizes the manuscript by section. If you have no clue how to do this, please try Overleaf. Format your preprint as though it was the only version of your report that people will ever read. Increasingly that’s the truth anyway.
In summary, I continue to think that eLife offers a great service to discerning authors and readers, and that its focus on open-access peer review points the way to a better future for scientific publishing.
I much enjoyed this book: “Models of the mind” by Grace Lindsay, an account of the uses of theory and mathematics in brain science. The book addresses the general reader, equations are mostly relegated to an appendix, and there are copious citations of technical literature as well. Obviously, conveying mathematics without equations is a challenge, and Lindsay handles it very well, finding many effective metaphors where needed. This all reflects her deep mastery of the material.
Beyond the target audience of curious laypeople, I also recommend this book to professors and students in mathematical neuroscience. For one, the ten chapters 2-11 would make a perfectly reasonable topic list for a 10-week course. While the course lectures and problem sets will focus on technical content and practice, Lindsay’s accompanying chapter can give a historical account of how the ideas evolved to our present understanding. When I try to do this in class, I always feel pressed for time, and the results aren’t satisfying. This book would make a great companion to a technical lecture series.
Lindsay’s last chapter is dedicated to “grand unifying theories” of the brain. She covers three attempts at such a theory, and politely but unmistakably dismisses them. One is “demonstrably wrong” and the other two are not even wrong. To find out what theories these are you’ll have to read the book; calling them out by name here would already give them too much credit. Lindsay concludes that brains are so “dense and messy” that they don’t lend themselves to physics-style theories that boil everything down to simple principles. On the other hand, her opening chapter makes the case that mathematics is the only language that can ultimately deal with the complexity of the brain. There’s a tension here regarding the future of mathematics in neuroscience.
This contrast between the rigor of mathematics and the squishiness of brains contributes to what I think is a continuing reluctance of many brain scientists to engage with quantitative methods. Here I’m reminded of one of the few question marks I jotted in the margins of the book. Lindsay writes that early in his career “[David] Hubel was actually quite interested in mathematics and physics”. If so, he certainly changed his mind later on. I have heard Hubel say that he “never had to use an equation after high school”. And he liked to ridicule quantitative measurement in neuroscience as “measuring the thickness of blades of grass”. Torsten Wiesel was similarly dismissive of mathematical approaches. I recall an editorial board meeting of the journal Network, which the chief editor, Joe Atick, had organized in a conference room at the Rockefeller University. In mid meeting, Torsten Wiesel – who was president of Rockefeller at the time – popped in, said hello, asked what the meeting was about, then delivered a monologue on how computational neuroscience will never make a useful contribution, and left abruptly. Whatever one thinks of these opinions, Hubel and Wiesel do have a Nobel Prize, as do many other experimenters, and theoretical neuroscientists don’t.
One can argue that Hubel and Wiesel really did not need mathematics to report the remarkable phenomena they discovered. But connecting those phenomena with their causes and consequences does require math. The little napkin sketch of simple cell receptive fields made from LGN neurons is cute but not convincing; it needs to be translated into a model before one can test the idea. Similarly, one can make a hand-waving argument that line detector neurons are useful for downstream visual processing, but understanding the reason is an entirely different matter. Unfortunately our discipline today still values isolated qualitative reports of phenomena. Most of the celebrated articles in glossy journals remain as singular contributions. No-one builds on them, hardly anyone tries to replicate them (and the rare attempts often don’t go well). Meanwhile the accompanying editorials celebrate the “tour-de-force” achievement that delivers a “quantum leap in our understanding”1.
We should resign ourselves to the recognition that no single research paper will pull the veil of ignorance from our eyes and reveal the workings of the brain. The only hope lies in integrating results that come from many researchers with diverse complementary approaches. And one can piece these results together only if they are reported with some quantitative precision2 and fit into an overall mathematical model. It is to enable this basic building of a scientific edifice that neuroscientists need to learn and use mathematics, not only for the pursuit of a grand unified theory.
Footnotes:
The next time you write this phrase, remember that a “quantum leap” is the smallest possible increase.
The next time you write a review article, please include some numbers about the magnitude of reported effects. Thank you!
Many years ago, in the early days of 2022, the dark specter of Covid finally lifted from American society. True, the disease was still killing Americans at a prodigious rate, equivalent to about 1 million a year. And most of those deaths were preventable, because other countries had done just that. What changed is that we decided to not worry about it anymore. Across the country and across the political spectrum, people agreed it was time to move on, to treat Covid like a seasonal flu, and to rescind any of the tedious mandates that had been imposed, like vaccinations or face coverings.
But the real transformation of American life that we all celebrate today happened in the immediate aftermath. Once we realized that a million deaths a year from a single cause of mortality was acceptable, we reconsidered all the irrational fears and regulations that had accumulated over the years. For example, Americans used to be asked for identification before boarding an airplane. Worse, they were forced to remove shoes, expose their bodies to X-rays, and let guards grope their genitals. All this was justified by the threat of terrorism. Now the most spectacular act of airplane terrorism known (at that time) killed about 3000 people in one day and then nothing happened for the next 20 years. Covid kills that number every single day! Being a rational people, we recognized the absurdity right away and rescinded all restrictions on air travel.
Next to fall were traffic regulations: At the time traffic killed about 41000 Americans a year, so it posed less than one twentieth of the risk from Covid. And what were Americans suffering just to retain such an absurdly low number? There were speed limits on the roads, restraints on the passengers called seat belts, and expensive safety designs built into cars. Worst of all: Americans were under a forced mandate to abstain from drinking while driving. All this is hard to understand: the average American was so much more likely to die while choking on a respirator than from a drunk driving accident. Needless to say we don’t live in fear anymore.
This was also the year when the utopian idea of gun control was finally abandoned. The number of gun-related deaths was already ridiculously low, only 45000 a year, barely a blip on the Covid scale. Then the drug laws were rationalized. Only 69000 americans died from opioid abuse that year, and some of that could be ascribed to Covid epidemic anyway. In any case, with our newfound acceptance of risk, no-one could justify regulating these drugs in any way. Other regulations soon followed. For example we used to have laws about how factories could pollute the air and water, again justified by some risk to life and health, which we now understand to be laughably low.
In retrospect it is hard for us to understand how our society had accumulated all these petty laws that restrict our freedom while protecting us from supposed risks. Covid finally unmasked these regulations as pure theater. No-one will contest that we are more free today than in the dark pre-Covid era. True, there are considerably fewer Americans alive than at the time, but some economists regard that as an added benefit. And, unlike in the old days, there aren’t any grandparents around to remind us of the old days.
tl;dr: Many scientific journals take a heavy hand when it comes to color choice in illustrations, with the noble goal of aiding colorblind readers. I argue that this policy is outdated, and in fact hurts the community it is intended to serve.
For example, the journal eLife (full disclosure: I contribute free services to this journal) instructs its authors: “When preparing figures, we recommend that authors follow the principles of Colour Universal Design (Masataka Okabe and Kei Ito, J*FLY), whereby colour schemes are chosen to ensure maximum accessibility for all types of colour vision.”
This policy has been superseded by the fact that everyone reads eLife on a computer display of some kind. And by now all computers offer a customized color transform for color-blind people. See these links for Windows, MacOS, and iOS. These color filters replace whatever colors are on the display with new colors that are best discriminated by someone with a color vision deficiency. Most importantly the filter can be optimized for the user’s specific form of color blindness. This service has been wildly popular with colorblind readers.
Under these conditions, following the eLife policy is in fact detrimental to colorblind readers. Recall that there is no single color palette that works best for all forms of color blindness. If the author adapts the palette to a particular 2-dimensional color space, say protanopia, that will be suboptimal for other readers. Here is an example using the document that the eLife policy cites for guidance (Okabe and Ito).
“Original” shows a classic red-green fluorescence micrograph. Below that is the color substitution recommended by Okabe & Ito: turn red into magenta. To the right are 3 images produced by the Mac OS filters for different forms of color-blindness (I photographed my display with my phone – crazy, I know). The version that Mac OS produces for protanopia is very close to what Okabe and Ito recommend. But note the other two versions for deuteranopia and tritanopia are quite different.
So following the recommended policy will favor protanopes but hinder deuteranopes. What is more, adapting the color palette to one of these abnormal color spaces will make it more difficult for the operating system to optimize the display for another space.
In conclusion the best policy for authors is to do what comes natural: choose colors that use the widest color gamut possible. Then let the user’s display device take over and implement that specific user’s preferences. By analogy, we don’t ask authors to write with 36-point font because some readers have poor vision. We know that the reader can turn up the magnification as suits her preference. The same is now true in color space.
This sub-genre of science fiction has made its way into mainstream science journals. I argue that the science is sketchy and the fiction is disappointing. Submit your own Biophyctional Journal abstracts in the comments!
Introduction
Science fiction rests on a compact between author and reader. The reader grants the author license to make some outrageous assumptions about the state of science and technology. In return the author spins an exciting yarn that may also hold some lessons about human nature. Recently a sub-genre of science fiction has made its entry into mainstream scientific journals. Again the authors ask readers to imagine that nature works very differently from what we know now, by factors of a million or up to 10 trillion. Then they speculate what might happen under those circumstances. Continue reading “Magnetofiction – A Reader’s Guide”→
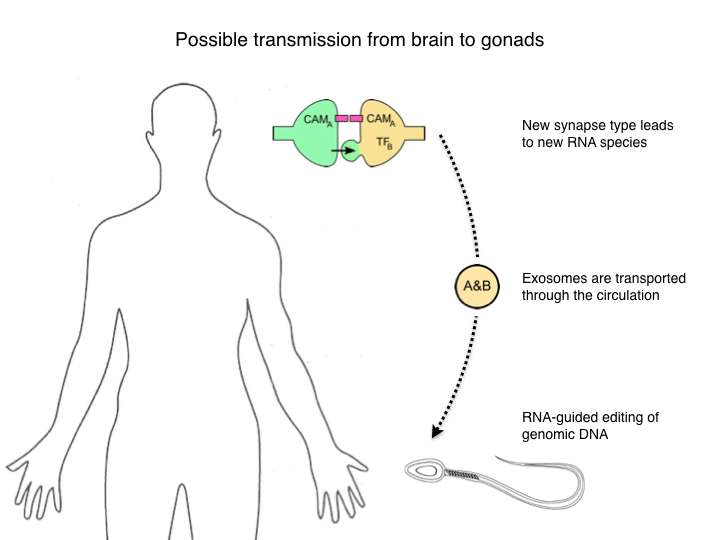
Preamble: In May 2018, HHMI Janelia hosted a wonderful conference on the evolution of neural circuits, organized by Albert Cardona, Melina Hale, and Gáspár Jékely. This is a transcript of a short talk I gave there. Caution: utter speculation!
Good evening. My name is Markus Meister. Many of you probably wonder what I am doing up here, and so do I. This started innocently enough when I wanted to attend this conference to learn about a different field. But the organizers asked for a poster abstract and I sheepishly complied. Then Albert Cardona unilaterally converted this poster to a talk, so now you know whom to blame. Let me also give two warnings. First, what I’ll present is pure speculation, there are no new results, only stringing together old results into questions. Fortunately it is timed to transition into the Apres-Ski part of the conference, so please consider this as part of the evening entertainment. Second I want to deliver a trigger warning. For those of you not at an American university, we professors are encouraged to warn our sensitive students when a lecture threatens to bring up a subject they might find offensive. The present talk will include notions reminiscent of Jean-Baptiste Lamarck, in particular his idea that acquired characters are inheritable.
This preprint presents new insights on visual processing in the retina, specifically how signals from rod photoreceptors are handled. Our visual system must operate over a huge range of light intensities, about 9 log units in the course of a day. In adaptation to this challenge the retina uses two kinds of photoreceptors: In the dimmest conditions only the sensitive rods are active, in the brightest conditions only the cones. In between the retina gradually switches from one input neuron to the other. However, even before the cones take over, the rod pathway undergoes substantial changes with increasing light level: the gain decreases and the speed of processing increases. This article challenges the prevailing notion of how those changes are accomplished.